Introducción
Los avances recientes en el conocimiento del genoma nos han permitido conocer la secuencia de los miles de genes que constituyen la vida. El ADN constituye la base hereditaria que, mediante las moléculas que transcribe de ARN, acaba dando lugar a las proteínas, moléculas responsables de la función biológica.
Cada proteína está formada por un número variable de aminoácidos, con una secuencia específica. El número, la clase y el orden de estos aminoácidos constituyen la denominada estructura primaria. No obstante, para realizar su función cada proteína requiere de una organización más compleja, una estructura tridimensional condicionada por las interacciones en la propia molécula denominada conformación o estructura nativa, caracterizada por un alto grado de estabilidad termodinámica.
Uno de los principales objetivos de la biología molecular ha sido y es la determinación de la función de las proteínas. Así mismo, uno de los fundamentos de la biología molecular establece que funciones similares en las proteínas están determinadas por estructuras similares y, por consiguiente, la función biológica de las proteínas está determinada por su estructura tridimensional. A su vez la estructura tridimensional está fundamentalmente determinada por las propiedades bioquímicas de la estructura primaria subyacente, es decir, por la cadena lineal de aminoácidos que constituyen dicha proteína. Es por tanto dicha cadena lineal la que controla la estructura tridimensional de las proteínas y, por consiguiente, su función.
Cuando se obtiene o se determina
una nueva proteína, se la clasifica teniendo en cuenta la
similitud de su secuencia lineal de aminoácidos con
secuencias de proteínas cuya función ya se conoce.
Tradicionalmente, esta clasificación se ha llevado a cabo por
medio de algún tipo de técnica de comparación de cadenas, tal
como Programación Dinámica,
donde dos secuencias son alineadas y se determina el coste del
alineamiento, sirviendo éste de base para la clasificación.
Este tipo de técnicas son apropiadas para secuencias
estrechamente relacionadas, los denominados homólogos
cercanos, que codifican funciones biológicas básicas. Sin
embargo cuanto más abstractas son las funciones biológicas de
interés, más débiles son las similitudes entre las secuencias
que definen a las proteínas, pero son muchas veces
precisamente éstos homólogos remotos los que resultan de más
interés para la biología molecular.
Para la clasificación de secuencias homólogas remotas, en lugar de Programación Dinámica se utilizan modelos probabilísticos de las familias de proteínas. Aunque estos modelos mejoran de forma significativa a las aproximaciones más tradicionales como Programación Dinámica, el problema de la clasificación de homólogos remotos está aún lejos de ser resuelto. Los modelos probabilísticos actuales sufren serios problemas impidiendo un avance mayor en el campo de la detección de homólogos remotos. Como ejemplo, se necesitan grandes conjuntos de entrenamiento para establecer buenos modelos, pero sin embargo para muchas familias de proteínas sólo disponemos de unas pocas secuencias. Como consecuencia de esto no se tiene claro, por ejemplo, la función de alrededor de un 40% de las proteínas humanas aunque hay fuertes sospechas de que buena parte de estas proteínas son en realidad homólogos distantes. Podemos concluir pues que son muy necesarios mejores modelos probabilísticos de las familias de proteínas, siendo éste un tema al que, de hecho, la comunidad científica le está dedicando un gran esfuerzo, constituyendo uno de los objetivos principales de este proyecto.
Una de las líneas que se ha empezado a explorar para avanzar en este aspecto es la representación de la secuencia de aminoácidos que constituyen la proteína como una señal (es decir, representando algún tipo de medida bioquímica dependiente de la posición particular dentro de la secuencia) frente al tradicional y abrumadoramente mayoritario tratamiento de la cadena de aminoácidos como una cadena simbólica constituida por letras de un alfabeto de 20 elementos donde cada letra representa a un aminoácido. La representación de la proteína como una señal permite que las numerosas y potentes herramientas del campo del Procesado de Señal se puedan aplicar al análisis y caracterización de las proteínas, posibilitando la mejora de los modelos probabilísticos. Aunque en los últimos años han aparecido algunos trabajos en la comunidad científica que van en esta línea, las aportaciones son aun pocas y el campo que queda por explorar es todavía amplio. Es en esta línea de investigación en la que pretendemos ahondar con este proyecto.
Objetivos del proyecto
La finalidad principal del presente proyecto es el desarrollo de mejores
técnicas computacionales de caracterización y clasificación de
proteínas a partir de su estructura primaria, mediante la
incorporación de herramientas propias del procesado digital de
señal y, en particular, la construcción de mejores modelos
computacionales para la detección y clasificación de proteínas
homólogas. Esto es de interés en muchos ámbitos de la biología
y particularmente para la industria farmacéutica.
Para ello, en lugar de usar la representación simbólica de la cadena de aminoácidos que constituye una proteína como se hace habitualmente, proponemos una nueva representación numérica para la cadena de aminoácidos que se construya incorporando las dependencias entre aminoácidos que aparecen reflejadas en las matrices de sustitución que se utilizan en las técnicas de alineamiento de cadenas simbólicas de aminoácidos. Este nuevo tipo de representación que proponemos genera una señal por cada aminoácido quedando cada proteína caracterizada por 20 señales.
Partiendo de dicha representación y basándonos en el modelo de reconocimiento resonante(Resonant Recognition Model, RRM), se obtendrá una caracterización de cada familia de proteínas mediante un conjunto de espectros de consenso y sus frecuencias características, desarrollándose posteriormente un clasificador basado en dicho modelo. De forma complementaria se desarrollará un clasificador más clásico basado en modelado probabilístico mediante Modelos Ocultos de Markov, que tenga como entradas vectores de características derivados de la representación en veinte señales propuesta para cada proteína. Se compararán ambos y se estudiará la posibilidad de combinarlos.
Finalmente a partir de la caracterización de las proteínas obtenida mediante el modelo RRM (complementada con el modelado estadístico desarrollado) pretendemos extraer información de carácter biológico a partir del mismo aplicando técnicas de filtrado digital de señal combinadas con conocimiento de carácter bioquímico.
Impacto
La información codificada en
la secuencia de aminoácidos, total o parcial, de una proteína
determina su conformación estructural y por tanto su función
biológica. La finalidad principal del presente proyecto es el
desarrollo de mejores técnicas computacionales de
clasificación y caracterización de proteínas
mediante la incorporación de herramientas propias del
procesado digital de señal y, en particular, la construcción
de mejores modelos computacionales para la detección y
clasificación de proteínas homólogas.
Esto nos permitiría identificar en proteínas, en las que ya se ha descrito su secuencia de aminoácidos, pero cuya función desconocemos, la conformación de la totalidad de la cadena, o segmentos de la cadena, a las que pudiera adscribirse una determinada función biológica.
Así mismo la comparación de proteínas de distintas especies nos podría dar claves sobre los procesos de la evolución y ayudar en la construcción de árboles filogenéticos. Pero además, la detección de nuevos miembros de ciertas familias de proteínas es especialmente interesante desde el punto de vista farmacéutico. En los últimos años la incorporación de herramientas computacionales en el proceso de descubrimiento de nuevos medicamentos está acortando los tiempos de dicho proceso y reduciendo su coste total. Además permite explorar y extraer nuevo conocimiento a partir de la ingente cantidad de datos que se están generando últimamente en el campo de la genómica y la proteómica: es la farmacogenética. En la figura siguiente se muestra un esquema del proceso de descubrimiento de un nuevo medicamento.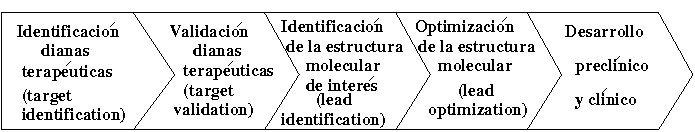
Aunque en los cuatro primeros pasos de dicho
proceso se están utilizando cada vez en mayor medida técnicas
computacionales que complementan a los métodos tradicionales
utilizados por la industria farmacéutica, es fundamentalmente
en el primer paso, el correspondiente a la identificación de
nuevas dianas terapéuticas, donde las técnicas que se proponen
en este proyecto tendrían más utilidad. La identificación de
nuevas dianas terapéuticas se realiza mediante una búsqueda
sistemática de proteínas (en su mayor parte) que realizan
funciones relacionadas. Así se analizan genomas completos con
respecto a ciertas familias de proteínas de interés
(relevantes desde un punto de vista farmacológico) en busca de
posibles dianas (screening). Una mejora en las técnicas de
análisis y modelado computacional de familias de proteínas,
como las que se proponen en el presente proyecto, haría más
efectiva la búsqueda inicial de dianas terapéuticas, pudiendo
ahorrarle tiempo y dinero a las empresas farmacéuticas
El establecimiento del Parque Tecnológico de Ciencias de la Salud (PTS-Granada, http://www.ptsgranada.com/) en el entorno de la ciudad de Granada está potenciando el interés por este tipo de investigaciones y facilitando la transferencia de tecnología generada por equipos de investigación de la Universidad de Granada a la empresa. Así, actualmente, en dicho campus se encuentran, el Centro de Investigación Biomédica, en el que se sitúan los Institutos de Investigación de Biotecnología, Neurociencias, Nutrición y Tecnología de los Alimentos, así como el de Biopatología y Medicina Regenerativa, además del Banco Andaluz de Células Madre, BACM; el Centro de Excelencia para la Investigación en Medicamentos Innovadores en Andalucía y el Centro de Genómica e Investigación Oncológica (GENyO), entre otros. También se encuentran centros de Desarrollo empresarial como son el Centro Europeo de Empresas e Innovación-BIC Granada, así como se encuentran implantadas empresas de referencia internacional, contando para ello con infraestructuras propias, como Neuron Bio o Laboratorios Farmacéuticos ROVI, entre otros. Otras empresas se encuentran asociadas al PTS, entre las que destacamos las del sector Bioinformático y que pueden consultarse en http://vicpts.ugr.es/pages/empresas/index#__doku_sector_bioinformatica.